- Home
- Introduction
- Downloads
- Example
-
User Guide
Index
 System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
- Screenshot
- Java Dev
- Plug-ins
- Visitors
25 Jan 2012:: We now have a new download site: www.OmicsExpress.com
25 May 2011:: SVA is published in Bioinformatics.
21 Mar 2011:: SVA V1.10 is released: [1]Supports GRCh build 37/hg19; [2] Supports user annotation track in GFF3 or BED formats.
9 Sep 2010:: The characterization of twenty sequenced human genomes. [Article]
12 Jul 2010:: LabCorp Launches Interleukin 28B Polymorphism (IL28B) Genotype Test to Support Individualized Treatment Decisions for Patients with Hepatitis C Viral Infection.
17 Jun 2010:: Causal variants for metachondromatosis are identified.
[Article] [SVA screenshot]
[GenomeWeb: The Daily Scan]
[Article] [SVA screenshot]
{kind=link}
[GenomeWeb: The Daily Scan]
18 Mar 2010:: SVA 1.02[beta] is released.
11 Mar 2010:: SVA 1.01[beta] is released with a command line tool.
8 Mar 2010:: A lite evaluation edition is released for Windows. Play with it on your laptop!
23 Jan 2010:: SVA 1.00[beta] is released.

An example project
The standard release of SVA provides an annotated example project. This example project is located at: [YOUR SVA DIR]/examples/X/cns.chrX.gsap .
|
A note on the example project: To reduce the size of the example project for your accessing convenience, this example contains only part (chromosome X) of the real data generated from a project including 5 patients with type A hemophilia patients resistant to HIV infection and 5 control genomes. This project is funded by the The Bill & Melinda Gates Foundation to David Goldstein at Duke University Center for Human Genome Variation . To further reduce the size of the example project, the coverage and quality score datasets (namely, the '.bco' files) were restricted to only one file (example.X.bco). The SVA .bco file contains the chromosome-wise read-depth and quality scores across the whole genome for each individual. Therefore this example project is only for demonstration and instruction purposes, although all the variant data together with their individual variant quality scores (other than the chromosome-wise coverage) are real. So please do not be surprised to see that the coverage plot in SVA Genome Browser displays the same coverage and quality scores for all the 10 subjects - this was intentionally done to reduce the project file size. In your own project, the .bco file should be separate for each chromosome and for each individual. |
Loading the example project
|
System requirements for loading the example project
|
Loading Steps
Click on the menu "File->Open an example project", you will see a dialog like this:

Click on "Load", you will see a project builder like this:
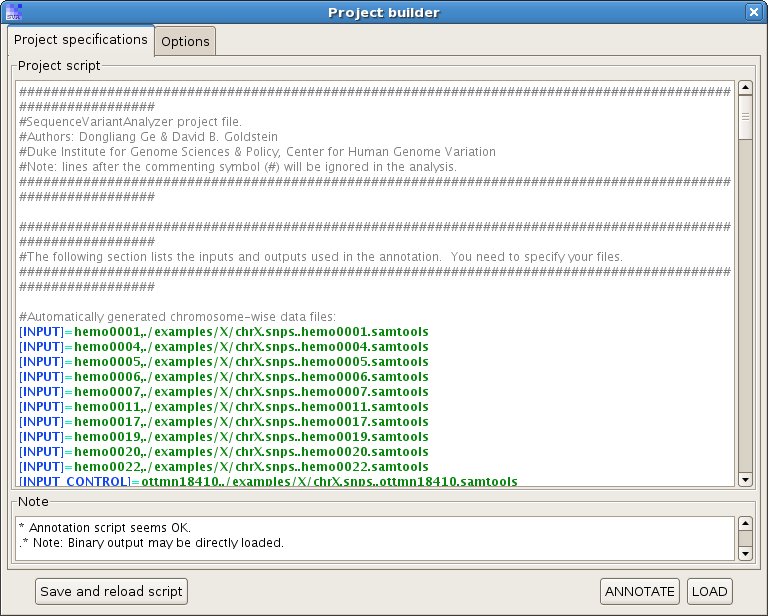
Check the option "Start up genome browser after loading the project", and hit "LOAD".
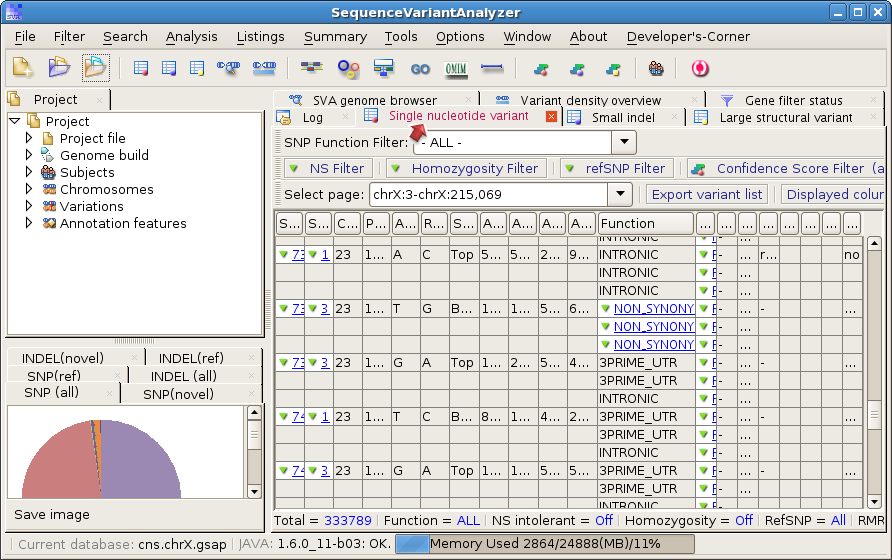
You should be able to see the variant list like this:
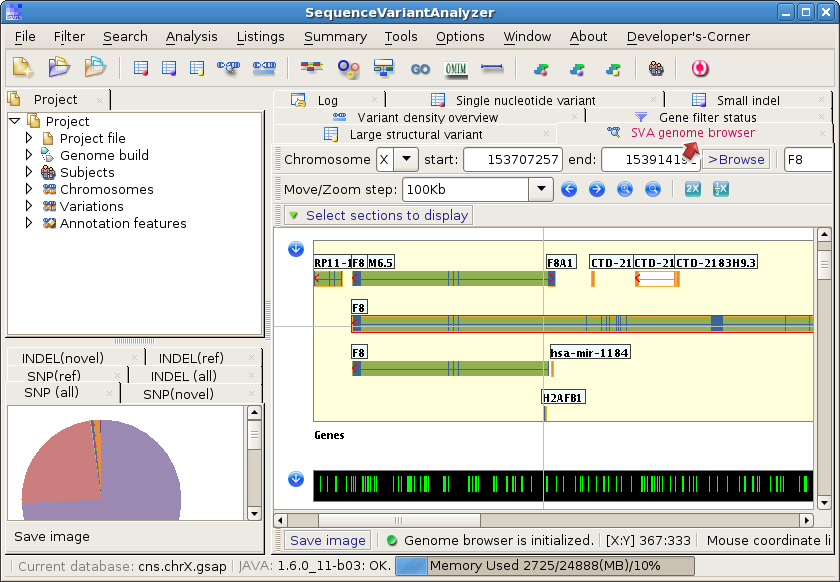
and search genes in the Genome Browser panel:
Feel free to play with the different functions with the example project. But if you want to create your own project and familiarize yourself with all the other functions, you probably need to go through the remaining documentations of SVA.
| Visits: |
© 2011
© 2011