- Home
- Introduction
- Downloads
- Example
-
User Guide
Index
 System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
- Screenshot
- Java Dev
- Plug-ins
- Visitors
Using this software
0. For impatient users1. Data inputs
2. Create a project
3. Annotate a project
4. Filter for quality scores
5. Main user interface
6. SVA genome browser
7. SVA tables
8. Selecting genes or regions
9. Analysis
10. Exome or targeted capture sequencing
A command line tool
FAQ
Requests and discussions

Annotating a project
After you create or open a project, click on button "ANNOTATE" in the "Project Builder" dialog to perform the main function annotation.
Again, I feel necessary to mention this important note one more time - sorry for being a nagger:
|
Important note: The default SVA release comes with a supporting database based on human reference sequence build 36 and Ensembl release 50_36l (June 2008 version). So it supports annotation and analysis consistent with that build. All the following steps, including the alignment processes upstream to this discussion, should also be based on human reference sequence build 36. The supporting database of SVA can be updated to newer builds. But the default SVA release is with build 36. We will release newer versions of supporting databases in the future. In any cases, the SVA supporting database version should be consistent with your alignment process. Otherwise, annotation will simply generate wrong results. The default SVA release is build 36. |
What does this process do?
This annotation process does three things:
|
1. This process will load the genetic variants specified in your SVA project .prj file. This process will compare the genetic variants across individual genomes and merge the same genetic variant records across individuals into one record including subject details- that is, SVA manages the genetic variant and subject data in a "variant-major mode" rather than a "subject-major mode"; 2. This process will annotate the loaded variants for their possible biological functions (defined here) ; 3. This process will order the loaded variants first by chromosome, then by coordinates, |
Sometimes, there will be a log file called "[YOUR SVA PROJECT FILE].exception.log" generated in your project folder (see figure below). This file saves all the genetic variant records in an unrecognizable format to SVA. If you include contigs not yet mapped to a chromosome in your reference sequence database when performing the alignment, those genetic variant mapped to contigs will, for example, be included in this file and will not be annotated for genomic functional context.
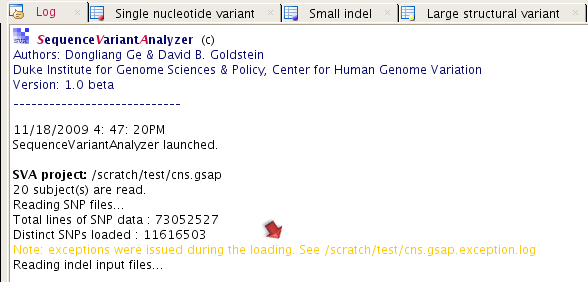
The function annotation may take 3-4 hours to complete for around 10 million variants, based on the computer we have been using. After the annotation is done, you will then be able to summarize and analyze the annotated data.
| Visits: |
© 2011
© 2011