- Home
- Introduction
- Downloads
- Example
-
User Guide
Index
 System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
- Screenshot
- Java Dev
- Plug-ins
- Visitors
Using this software
0. For impatient users1. Data inputs
2. Create a project
3. Annotate a project
4. Filter for quality scores
5. Main user interface
6. SVA genome browser
7. SVA tables
8. Selecting genes or regions
9. Analysis
10. Exome or targeted capture sequencing
A command line tool
FAQ
Requests and discussions

A fast kick-off for impatient users
The standard release of SVA provides an annotated example project that you can have a quick taste before you dive into this documentation and create your own projects.
This example project is located at: [YOUR SVA DIR]/examples/X/cns.chrX.gsap .
|
System requirements for loading the example project
|
Loading Steps
Step 1: Loading the example project
You need about 3GB of RAM to load the example project with its genome browser data. You need about 0.8GB RAM to load the example project without its genome browser data, in which case you will be able to list the annotated variants, but cannot search genes in the SVA Genome Browser panel, or perform following analysis.
Click on the menu "File->Open an example project", you will see a dialog like this:

Click on "Load", you will see a project builder like this:
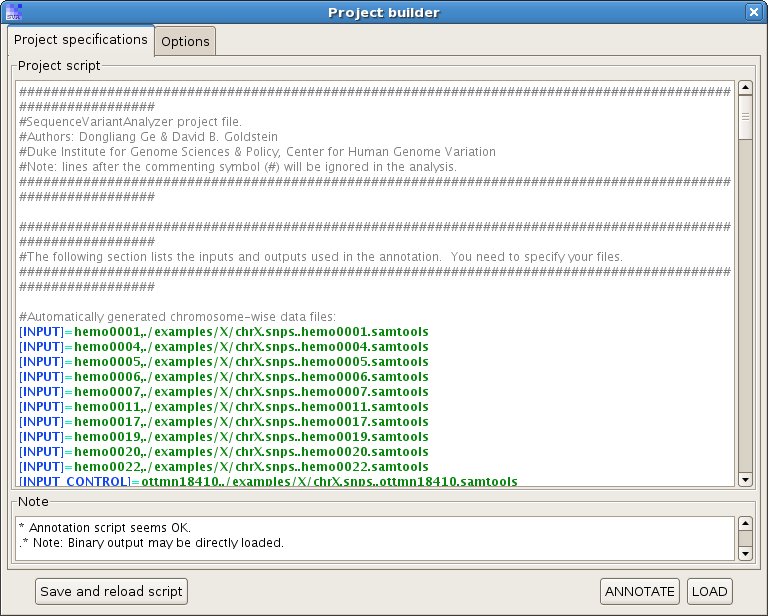
Check the option "Start up genome browser after loading the project", and hit "LOAD".
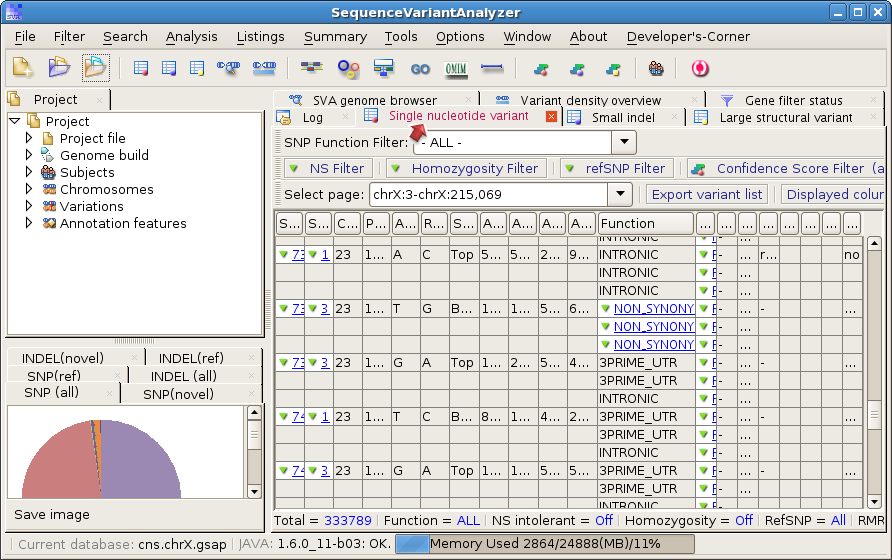
You should be able to see the variant list like this:
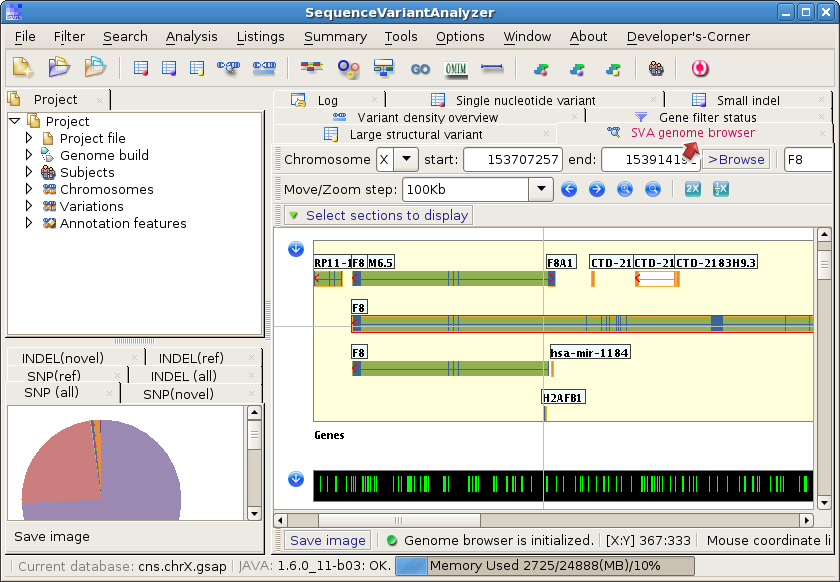
and search genes in the Genome Browser panel:
Step 2: Performing a "Gene Prioritization" analysis
The first analysis you may want to play within the example project is "Gene Prioritization". This analysis ranks genes by the number of cases carrying homozygous protein-truncating genetic variants that do not appear in control genomes. Please note by this definition different variants in the same gene are allowed and counted. In other words, this analysis tries to answer this simple question: is there any enrichment of protein-truncating variants in any gene in the cases, but none of these variants present in the control genomes?
To perform this analysis, click menu "Analysis -> Gene prioritization". You will see a parameter dialog like this:
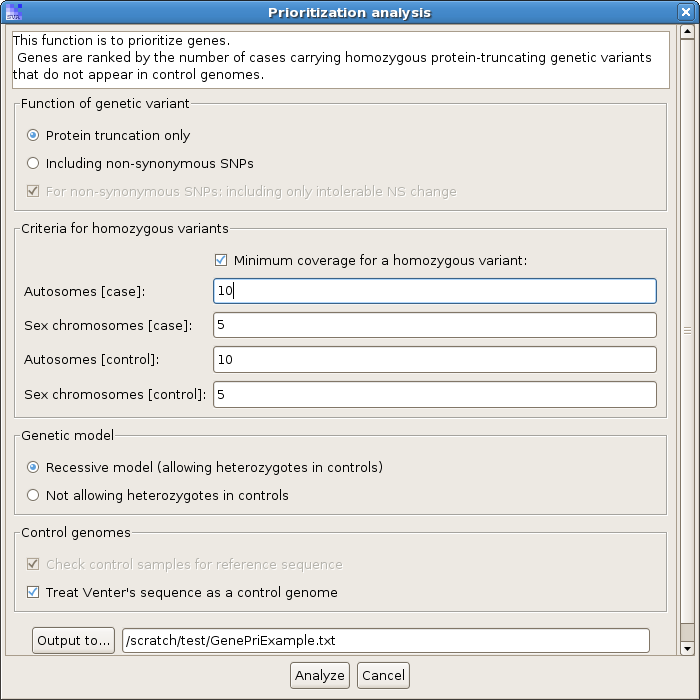
Input the same parameters, or play with the parameters if you want, and you will see a result like this:
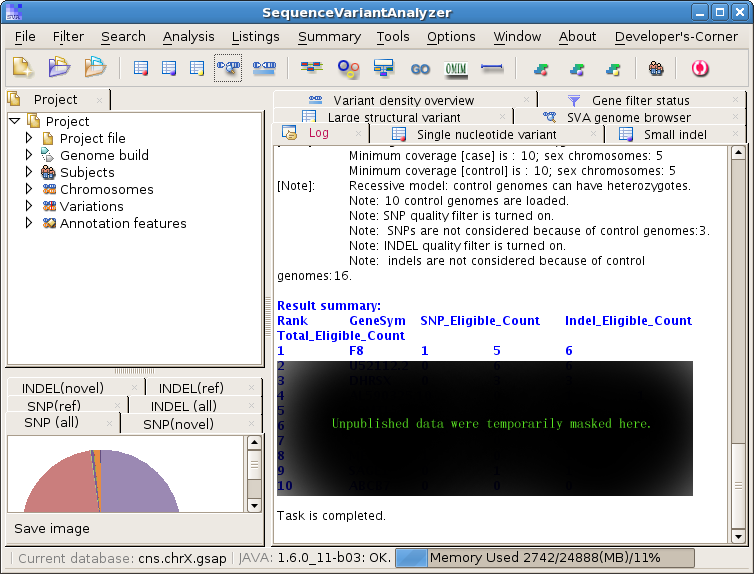
As you see from the result screen, the Factor VIII ( F8 ) gene is ranked as the 1st target of enrichment in the case genomes. This ranking is based on several different protein-truncating SNPs and INDELs. This ranking is satisfying since the case genomes in our example project are type A hemophilia patients and we know that F8 mutations lead to hemophilia A. This ranking also holds true when we run this analysis on our complete dataset including all the chromosomes.
This analysis is one of the several methods that we are currently developing. Feel free to play with the different functions with the example project. But if you want to create your own project and familiarize yourself with all the other functions, you probably need to go through the remaining documentations of SVA.
| Visits: |
© 2011
© 2011